Overview
The KnockOffGWAS pipeline was constructed by adapting the example given by the author(s) of KnockOffGWAS. This example provided all the necessary files to perform KnockOffGWAS for this example but was not suitable for general use. The following list is a description of the new pipeline:
The new pipeline is entirely contained within the directory new_knockoffgwas_pipeline.
The scripts have been updated to include parameters for the name of data files rather than be hard-coded with filenames.
There are many data files (such as IBD files) that are required before the KnockOffGWAS analysis can be ran. Therefore there have been many scripts and programs added to the pipeline to perform this required data preparation.
The new parts to this pipeline are stored in the new_bits directory.
In principle, one should be able to run the scripts run_pre_create_map_files.sh, run_pre_phasing.sh, run_pre_ibd.sh and run_knockoff_gwas.sh in order to perform KnockOffGWAS (in this order).
To view the results and produce plots the the Shiny-R app, app.R, in the visualisation directory can be run.
To remove all remaining temporary files produced during analysis the run_remove_temporary_files.sh script can be ran. If you wish to rerun the analysis for any reason you will need to rerun the data preprocessing scripts if you remove the temporary files.
The analysis is computationally intensive for real datasets and will need to be ran on an High Performance Computing (HPC) machine. With this in mind, this webpage describes the process of performing the analysis of a real dataset on an HPC machine.
By following the steps on this website it should be possible to perform KnockOffGWAS on your dataset. As an example of using this pipeline, the directory pbc_analysis contains additional scripts to run analysis for this data on an HPC machine. Hopefully, with this pipeline, guide and worked example it should be as straightforward as it can be to perform KnockOffGWAS considering the many programs and different data formats involved.
A containerised version is also provided to facilitate deployment across different systems. See Pipeline Container.
The following is the structure and contents of the new_knockoffgwas_pipeline directory:
new_knockoffgwas_pipeline
├── knockoffgwas_pipeline
│ ├── analyze.sh
│ ├── knockoffgwas
│ │ ├── module_1_partition.sh
│ │ ├── module_2b_knockoffs_gof.sh
│ │ ├── module_2_knockoffs.sh
│ │ ├── module_3_statistics.sh
│ │ ├── module_4_discover.sh
│ │ └── utils
│ │ ├── check_packages.R
│ │ ├── filter_stats.R
│ │ ├── knockoffs_gof.R
│ │ ├── lasso.R
│ │ ├── list_original.R
│ │ ├── make_FBM.R
│ │ ├── merge_chromosomes.sh
│ │ └── partition.R
│ ├── misc
│ │ ├── download_annotations.sh
│ │ ├── lmm.sh
│ │ ├── makedata.sh
│ │ ├── make_genetic.R
│ │ ├── make_phenotypes.R
│ │ ├── spinner.sh
│ │ └── summarise_lmm.R
│ ├── new_bits
│ │ ├── convert_fam_format.R
│ │ ├── convert_ibd_format3.R
│ │ ├── convert_ped_file.R
│ │ ├── convert_sample_format.R
│ │ ├── filter_ibd_file.R
│ │ ├── filter_mapping_file.py
│ │ ├── filter_mapping_file.R
│ │ ├── interpolate_genetic_map.R
│ │ ├── interpolate_loci.py
│ │ ├── interpolate_loci.R
│ │ ├── make_phenotype_file_from_fam.R
│ │ ├── make_qc_files.R
│ │ ├── parameter_estimation.py
│ │ ├── parameter_estimation.R
│ │ ├── phase_common_static
│ │ └── RaPID_v.1.7
│ ├── snpknock2
│ │ └── bin
│ │ └── snpknock2
│ ├── visualization
│ │ ├── app.R
│ │ ├── check_packages.R
│ │ ├── test_plot.R
│ │ ├── utils_clumping.R
│ │ ├── utils_fdp.R
│ │ ├── utils_manhattan.R
│ │ ├── utils_plotting.R
│ │ ├── utils_shiny.R
│ │ └── www
│ │ └── theme.css
│ └── visualize.sh
├── run_knockoff_gwas.sh
├── run_pre_knockoff_gwas.sh
└── run_remove_temporary_files.sh
Below is a flow diagram of the KnockOffGWAS pipeline.
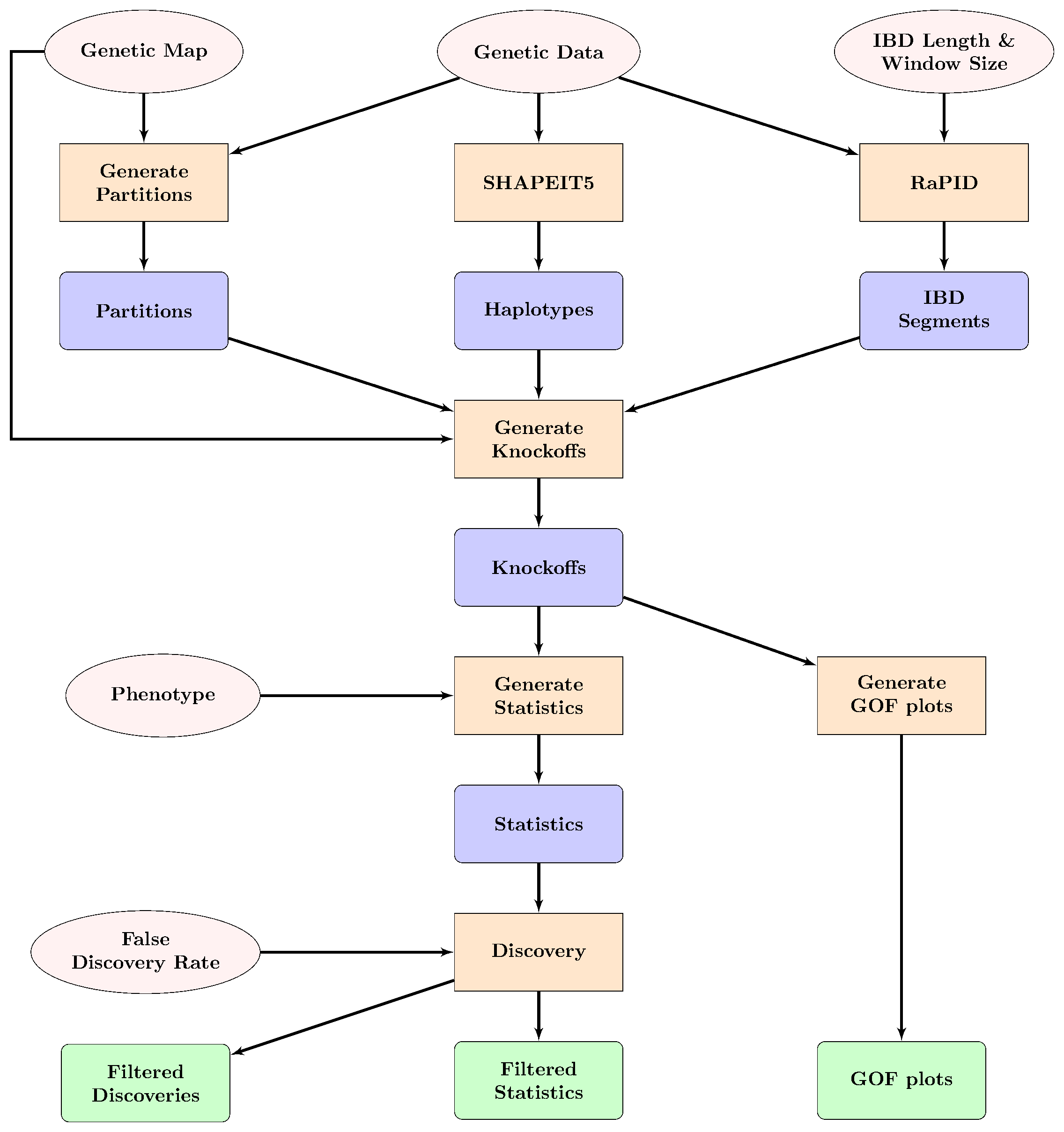
Flow diagram of the KnockOfGWAS pipeline. The pink ovals show input data and parameters, the orange squares show data processing steps, blue rounded squares show intermediate data and the green rounded squares show final output.